


Restoring Endothelial Homeostasis for Alzheimer’s Therapy: Mechanistic and Translational Insights from Multivalent Modulation of Blood–Brain Barrier Transport
Executive Summary
Recent findings demonstrate that the blood–brain barrier (BBB) can be reprogrammed from a dysfunctional conduit into a therapeutic target for Alzheimer’s disease (AD). Chen et al. (2025) report that mid-avidity, multivalent nanocarriers targeting the low-density lipoprotein receptor–related protein 1 (LRP1) restore endothelial transport dynamics, rapidly clear amyloid-β (Aβ) from the brain, and yield sustained cognitive recovery in murine models. The study highlights endothelial homeostasis—not plaque dissolution—as a viable disease-modifying axis. For industry leaders, this approach reframes neurovascular biology as a new therapeutic domain with implications for molecular design, biomarker development, and regulatory assessment of BBB-directed drugs.
Abstract
The vascular contribution to neurodegeneration has long been recognized, yet practical means of therapeutically repairing the blood–brain barrier (BBB) have remained elusive. In Alzheimer’s disease (AD), BBB dysfunction reduces Aβ efflux and promotes parenchymal deposition. Chen et al. (2025) introduce a supramolecular strategy leveraging multivalent polymersomes decorated with angiopep-2 ligands to modulate LRP1-dependent transcytosis. By tuning ligand avidity, the investigators stabilize a PACSIN2-linked tubular trafficking pathway, leading to rapid Aβ clearance, normalization of endothelial signatures, and enduring cognitive restoration in APP/PS1 mice. This white paper examines mechanistic underpinnings of LRP1 transport, contrasts the approach with antibody-based therapies, and discusses translational requirements for human application, including pharmacodynamic biomarkers, CMC constraints, and regulatory precedents.
Introduction
Alzheimer’s disease remains an area of high unmet medical need despite recent therapeutic approvals. Lecanemab and donanemab have validated Aβ reduction as a tractable clinical endpoint (van Dyck et al., 2023; Sims et al., 2023), yet limitations—including amyloid-related imaging abnormalities (ARIA), incomplete cognitive benefit, and dosing constraints—underscore the necessity for complementary mechanisms (Salloway et al., 2025). Mounting evidence positions BBB dysfunction as both an early event and a driver of neurodegeneration, particularly in carriers of the APOE4 allele (Montagne et al., 2020). Endothelial degradation of LRP1 and associated trafficking proteins impairs Aβ efflux, while neuroinflammation and oxidative stress exacerbate permeability defects (Storck et al., 2016; Zhao et al., 2015). The study by Chen et al. (2025) offers a mechanistically grounded approach to repair this transport axis.
Mechanistic Framework: LRP1-Mediated Transcytosis and Endothelial AvidityThe LRP1 clearance axis
LRP1 is a 600-kDa endocytic receptor expressed in brain endothelium and astrocytes. It mediates transcytosis of diverse ligands, including Aβ, apolipoprotein E complexes, and protease-inhibitor conjugates. Under physiological conditions, Aβ binds to LRP1 on the abluminal membrane and is trafficked toward the luminal surface via vesicular and tubular intermediates, cooperating with P-glycoprotein (ABCB1) and PICALM (Storck et al., 2018; Zhao et al., 2015).
During AD progression, oxidative stress and vascular inflammation internalize and degrade LRP1, diverting cargo into Rab5-positive endosomes and reducing efflux efficiency. This maladaptive routing contributes to perivascular amyloid deposition and cerebral amyloid angiopathy (CAA). Restoring tubular transcytosis therefore represents a direct intervention on the pathophysiological machinery of Aβ accumulation.
Avidity and trafficking fate
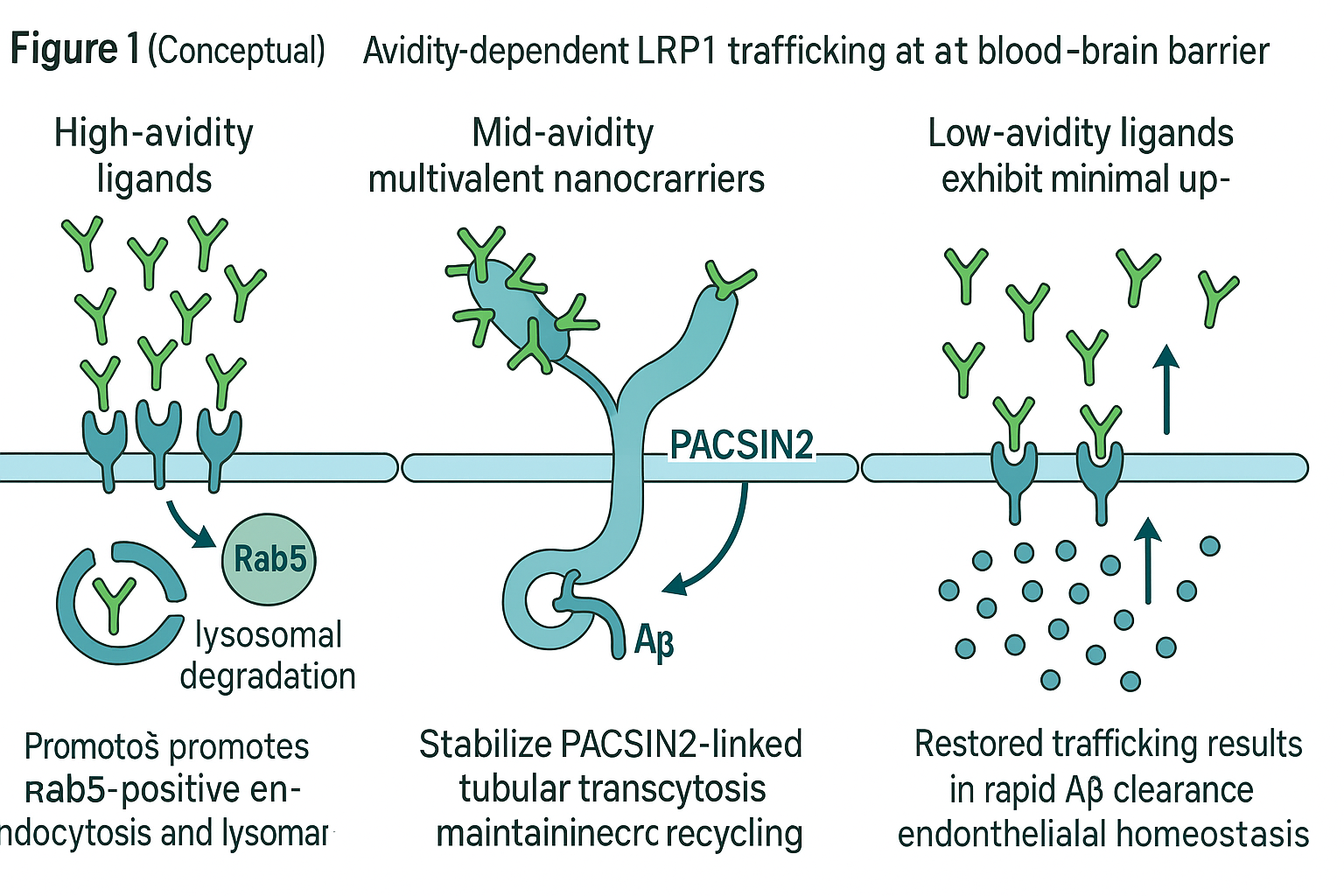
Ligand–receptor interactions at the BBB operate under an “avidity window”: too low an affinity fails to engage endocytosis, whereas excessive avidity causes receptor clustering and lysosomal degradation (Yu et al., 2011; Niewoehner et al., 2014). Figure 1 schematically illustrates how mid-avidity multivalency—approximately 40 ligands per 100 nm polymersome—stabilizes LRP1 within PACSIN2-positive tubular structures that support bidirectional transcytosis. In contrast, high-avidity binding diverts LRP1 to degradative compartments, further compromising efflux.
Experimental Overview: Chen et al. (2025)
Nanocarrier design and biophysical optimization
Chen et al. developed biodegradable polymersomes functionalized with variable densities of angiopep-2, a peptide ligand of LRP1. Dynamic light scattering confirmed a uniform hydrodynamic diameter of ~100 nm and narrow polydispersity. Surface decoration was modulated from 10 to 100 ligands per particle to map the avidity-response curve. Mid-avidity formulations (A40-POs) achieved maximal brain penetration and minimal lysosomal colocalization.
Pharmacodynamic and histological findings
In APP/PS1 mice, a single intravenous injection of A40-POs produced:
-
Aβ reduction of 41–50% within 2 h (post-ELISA and PET-CT quantification).
-
Plasma Aβ elevation (~8-fold), indicating increased brain-to-blood efflux.
-
Restoration of endothelial markers: LRP1–CD31 colocalization normalized; PACSIN2 expression increased, while Rab5 decreased.
-
Cognitive recovery: Morris Water Maze performance equaled wild-type levels for up to six months.
Transmission electron microscopy revealed re-emergence of tubular LRP1-positive structures. Proteomic analyses confirmed re-enrichment of transport proteins and suppression of inflammatory signatures, supporting the conclusion that nanocarrier exposure reprograms endothelial trafficking toward homeostasis.
Mechanistic proposition
The authors propose that mid-avidity engagement forms a “transportosome” complex linking LRP1, PACSIN2, and cytoskeletal scaffolds, allowing continuous receptor recycling rather than consumption. This mechanism contrasts with antibody-mediated Aβ extraction, which relies on peripheral sink effects or Fc-mediated plaque opsonization.
Translational and Clinical Implications
Orthogonality to antibody therapy
Unlike monoclonal antibodies that target extracellular Aβ aggregates, multivalent BBB modulation acts upstream by restoring physiological clearance. The mechanism may complement antibody therapy, potentially lowering required doses and mitigating ARIA through smoother amyloid efflux kinetics. Such combination strategies could be evaluated in controlled co-administration designs.
Biomarker development
Translational progression will depend on defining measurable surrogates of BBB repair:
-
Molecular biomarkers: Soluble LRP1, PACSIN2, and Rab5 levels in cerebrospinal fluid (CSF) or endothelial extracellular vesicles.
-
Imaging markers: Dynamic contrast-enhanced MRI to quantify BBB permeability; PET tracers for LRP1 or Aβ flux; arterial-spin-labeling perfusion metrics.
-
Peripheral correlates: Plasma Aβ^40/42 ratio and neurofilament light chain as indicators of neurovascular stress.
These endpoints could align with existing regulatory familiarity from antibody programs (FDA, 2023; Eisai, 2025).
Manufacturing and CMC considerations
Reproducible control of particle size, ligand density, and spatial distribution is critical. Each parameter directly influences avidity and trafficking fate. CMC protocols must ensure batch-to-batch stability of ligand presentation and polymer degradation rate. Analytical methods such as cryo-TEM and nanoparticle tracking analysis will be essential for release testing. Establishing structure–function correlations early will streamline regulatory review.
Clinical trial design
First-in-human studies will likely adopt a single-ascending-dose format emphasizing safety, vascular tolerance, and pharmacodynamic readouts. Inclusion criteria may stratify by APOE genotype and CAA burden, given differential BBB fragility (Montagne et al., 2020). Combination cohorts with antibody therapy could explore additive clearance without increasing ARIA incidence.
Challenges and Limitations
Species translation
Murine BBB characteristics differ substantially from humans in endothelial heterogeneity, shear stress exposure, and pericyte coverage. Humanized in vitro models and non-human primate studies under physiological flow are required to confirm transcytosis dynamics.
Complexity of LRP1 signaling
LRP1 influences both Aβ clearance and amyloid precursor protein (APP) processing. Overactivation may inadvertently enhance amyloidogenic pathways or disturb lipid metabolism (Van Gool et al., 2019). Dose optimization and longitudinal biomarker monitoring will therefore be critical.
Immunogenicity and biodistribution
Although polymersomes are non-antibody constructs, repetitive peptide presentation could elicit humoral responses over chronic dosing. Surface PEGylation and immune-stealth design may mitigate this risk. Additionally, systemic LRP1 expression in liver and vasculature necessitates biodistribution modeling to prevent off-target sequestration.
Regulatory novelty
While regulators have experience with gene therapies and antibodies, BBB-repair nanomedicines represent an emerging class. Early dialogue with authorities will be needed to define acceptable endpoints and comparators, particularly for hybrid mechanisms that influence both vascular and neural compartments.
Future Outlook
The Chen et al. (2025) study underscores a paradigm shift: the BBB is not merely an obstacle to drug delivery but a modifiable organ whose dysfunction drives neurodegeneration. The capacity to normalize endothelial trafficking introduces a new dimension to therapeutic strategy.
Future investigations may extend multivalent design to other receptor systems (e.g., transferrin receptor 1, insulin receptor) or apply artificial-intelligence optimization to avidity landscapes. Integration with gene-editing or RNA-based modalities could yield hybrid therapies that combine vectorized delivery with vascular modulation.
In the longer term, quantitative systems pharmacology models will be required to couple receptor kinetics, vascular transport, and amyloid dynamics, informing dose prediction and combination scheduling. Such efforts will clarify whether BBB repair can achieve disease modification comparable to or greater than current antibody programs.
Figure 1 (Conceptual)
Schematic overview of avidity-dependent LRP1 trafficking at the blood–brain barrier.
-
Left: High-avidity ligands cluster LRP1, promoting Rab5-positive endocytosis and lysosomal degradation.
-
Center: Mid-avidity multivalent nanocarriers stabilize PACSIN2-linked tubular transcytosis, maintaining receptor recycling and efficient Aβ efflux.
-
Right: Low-avidity ligands exhibit minimal uptake. Restored trafficking results in rapid Aβ clearance and endothelial homeostasis.
Conclusion
By targeting the mechanistic root of BBB failure rather than its consequences, multivalent modulation of LRP1 transport offers a mechanistically distinct route to cognitive recovery. The translational path will demand interdisciplinary collaboration among vascular biologists, nanotechnologists, clinicians, and regulatory scientists. If successfully implemented, BBB-repair therapeutics could redefine the treatment architecture of Alzheimer’s disease and related neurodegenerative disorders.
References
Chen, J., Xiang, P., Duro-Castano, A., Cai, H., Guo, B., Liu, X., Yu, Y., Lui, S., Luo, K., Ke, B., Ruiz-Pérez, L., Gong, Q., Tian, X., & Battaglia, G. (2025). Rapid amyloid-β clearance and cognitive recovery through multivalent modulation of blood–brain barrier transport. Signal Transduction and Targeted Therapy, 10, 331. https://doi.org/10.1038/s41392-025-02426-1
Doran, S. J., Hall, A., Gupta, Y., & Chappell, F. (2024). Risk factors in developing amyloid-related imaging abnormalities (ARIA). Frontiers in Radiology, 4, 1235013. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10834650/
Eisai Co., Ltd. (2025, April 16). Leqembi is the first medicine that slows progression of early Alzheimer’s disease authorized in the EU. https://www.eisai.com/news/2025/news202532.html
Hampel, H., Cummings, J., Blennow, K., & Lista, S. (2023). Amyloid-related imaging abnormalities (ARIA): Radiological, biological and clinical characteristics. Brain, 146(11), 4414–4432. https://academic.oup.com/brain/article/146/11/4414/7191051
Montagne, A., Nation, D. A., Sagare, A. P., et al. (2020). APOE4 leads to blood–brain barrier dysfunction predicting cognitive decline. Nature, 581, 71–76. https://www.nature.com/articles/s41586-020-2247-3
Niewoehner, J., Bohrmann, B., Collin, L., et al. (2014). Increased brain penetration and potency of a therapeutic antibody using a monovalent molecular shuttle. Neuron, 81(1), 49–60. https://core.ac.uk/download/pdf/82728521.pdf
Salloway, S., Mummery, C., Cummings, J., et al. (2025). Amyloid-related imaging abnormalities in clinical trials and practice. JAMA Neurology, 82(x), e1–e10. https://jamanetwork.com/journals/jamaneurology/fullarticle/2826606
Sims, J. R., Zimmer, J. A., Evans, C. D., et al. (2023). Donanemab in early symptomatic Alzheimer disease: TRAILBLAZER-ALZ 2. JAMA, 330(6), 512–527. https://pubmed.ncbi.nlm.nih.gov/37459141/
Storck, S. E., Meister, S., Nahrath, J., et al. (2016). Endothelial LRP1 transports amyloid-β1-42 across the BBB. Journal of Clinical Investigation, 126(1), 123–136. https://www.jci.org/articles/view/81108
Storck, S. E., Pietrzik, C. U., et al. (2018). Concerted Aβ clearance by LRP1 and ABCB1/P-gp is linked by PICALM. Brain, Behavior, and Immunity, 73, 21–33. https://pubmed.ncbi.nlm.nih.gov/30041013/
U.S. Food and Drug Administration. (2023, July 6). FDA converts novel Alzheimer’s disease treatment to traditional approval (Leqembi). https://www.fda.gov/news-events/press-announcements/fda-converts-novel-alzheimers-disease-treatment-traditional-approval
van Dyck, C. H., Swanson, C. J., Aisen, P., et al. (2023). Lecanemab in early Alzheimer’s disease. New England Journal of Medicine, 388, 9–21. https://www.nejm.org/doi/full/10.1056/NEJMoa2212948
Van Gool, B., Storck, S. E., Reekmans, S. M., et al. (2019). LRP1 has a predominant role in production over clearance of Aβ in a mouse model. Molecular Neurobiology, 56, 7234–7245. https://link.springer.com/article/10.1007/s12035-019-1594-2
Yu, Y. J., Zhang, Y., Kenrick, M., et al. (2011). Boosting brain uptake of a therapeutic antibody by reducing affinity for a transcytosis target. Science Translational Medicine, 3(84), 84ra44. https://pubmed.ncbi.nlm.nih.gov/21613623/
Zhao, Z., Sagare, A. P., Ma
Notes on sources and rights: The primary paper (Chen et al., 2025) is open access (CC BY 4.0), and specific quantitative and mechanistic details above are drawn from that article.
Primary Paper can be VIEWED HERE or ONLINE.